
Written by Mason Motal, Aaron Murski, CVA, Austin Grawe, and Patrick Speights
The coronavirus disease 2019 (COVID-19) is caused by the virus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and was first discovered in December 2019 in Wuhan, China. COVID-19 at the time was a new human virus causing respiratory illness and was very contagious, quickly spreading from person to person. Within a few months it was announced by the CDC as a global pandemic on March 11, 2020. The laboratory industry began leading at the forefront to combat the virus, and faced many unique, complex challenges. Hundreds of thousands of people in the United States were getting swabbed every day for COVID-19, and this caused labs to face severe supply shortages and for staffs to be overwhelmed with more tests than they could handle. Laboratory employees were working 24/7 around-the-clock to try to keep up with the high testing demand. Still, they were running out of essential products faster than manufacturers could restock them.
“If the demand keeps increasing like it’s been increasing over the last couple of weeks, the lab industry will never be able to keep up with it,” said Dr. Dwayne Breining, executive director of Northwell Health Laboratories. “I think the top priority is going to be mitigating the clinical spread of the virus by doing things we know work: things like social distancing, masking, and monitoring. That would allow you to slow it down enough so that not only the lab testing industry, but the entire medical system can catch up.” (6) (10)
Clinical Laboratory Improvement Amendments Overview
The Clinical Laboratory Improvement Amendments (CLIA) was established in 1988 to strengthen federal oversight of clinical laboratories and prohibit them from testing human specimens for diagnosis, prevention, treatment, or health assessment without a valid CLIA certificate. To become CLIA-certified labs must establish specific performance characteristics such as accuracy, quality, and reliability as imposed by the CLIA statute. According to the CDC, approximately 14 billion lab tests are ordered annually, and roughly 70 percent of today’s medical decisions are based on lab results. Therefore, CLIA ensures that patients and health care providers receive accurate, dependable test results.
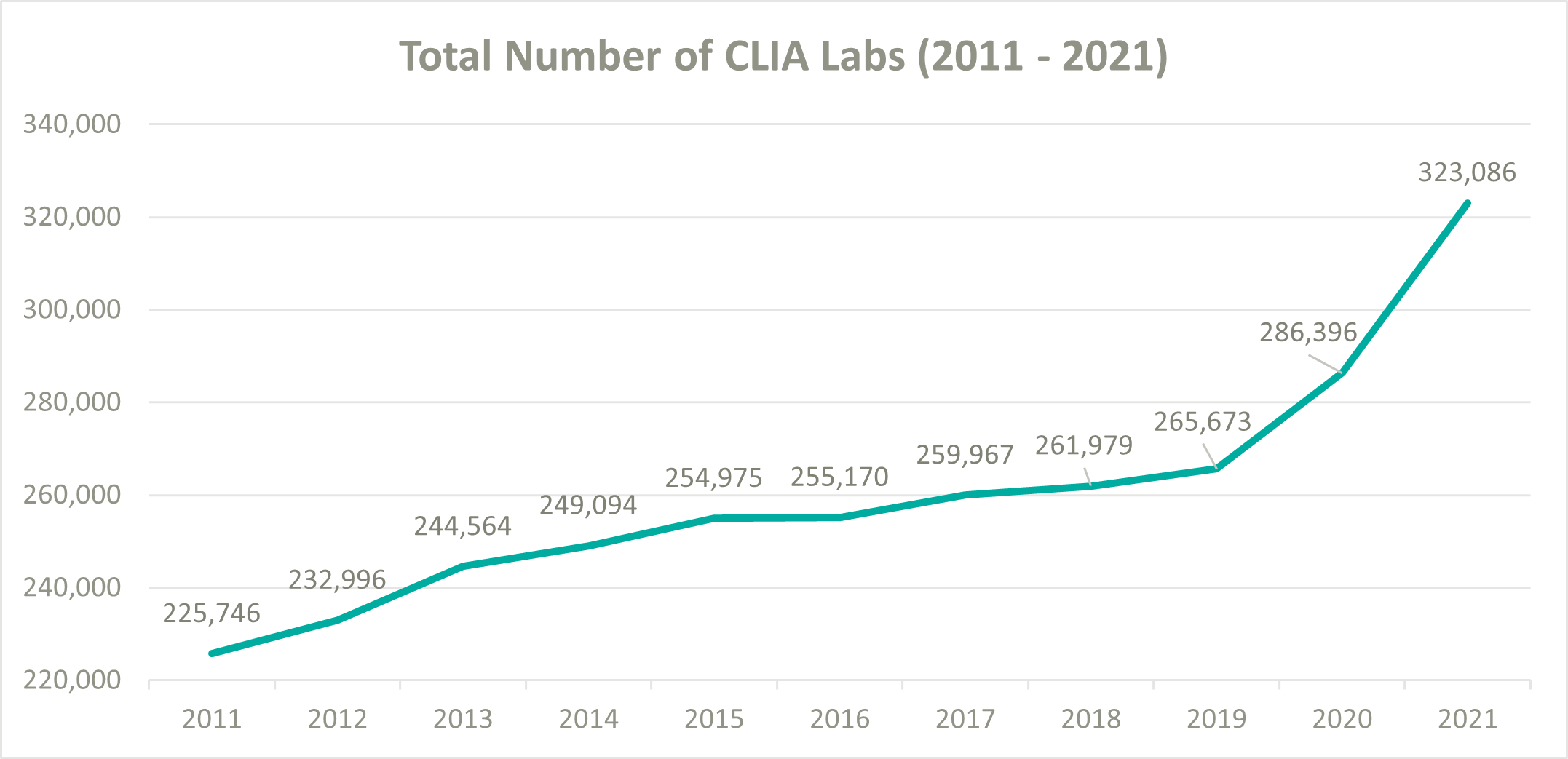
To help ease pressure on the lab industry during the onset of COVID-19, the Centers for Medicare and Medicaid Services (CMS) decided to loosen their CLIA policies and allowed laboratories wishing to perform COVID-19 tests to become CLIA certified and begin testing patients as quickly as possible. According to the CMS database, the total number of CLIA labs increased from 265,673 at the beginning of 2020 to 323,086 by the end of 2021, resulting in a 10.3% compound annual growth rate (CAGR). In comparison, from 2011 to 2019 the number of CLIA labs only grew from 225,746 to 265,673, a 2.1% CAGR. (5) (1) (7)
Laboratory-Developed Tests Overview
Typically, laboratory tests are developed by diagnostics manufacturers that produce test kits to sell to clinical labs. However, tests are also developed by labs that manufacture and use them in their own facilities, known as laboratory-developed tests (LDTs). LDTs are defined by the Food and Drug Administration (FDA) as an in vitro (in glass) diagnostic test that is manufactured by and used within a single laboratory. LDTs are commonly referred to as “in-house” or “home brew” tests. The FDA regulates the manufacturing of test kits by diagnostics manufacturers under its medical device authorities. Still, it exercises regulatory discretion to allow labs to use their LDTs pursuant to rules established under CLIA. CLIA’s analytical validation is limited to specific conditions, staff, equipment, and patient populations. Therefore, LDTs are reviewed during the routine biennial survey and are constantly monitored after the testing begins.
Furthermore, CLIA requirements are based on test complexity, therefore labs that develop LDTs must meet stricter requirements than standard labs. In contrast, the FDA’s premarket clearance and approval processes assess the analytical validity of a test system in much greater depth and scope. The FDA’s review of analytical validity is done prior to the marketing of the test system, as well as assessing clinical validity, which is part of the review focused on the safety and effectiveness of the test system. The CLIA and FDA regulatory systems differ in levels of focus, scope, and purpose; however, they are intended to be complementary to each other. (1) (2) (13)

LDTs Impacting the Lab Industry
When COVID-19 initially hit the U.S. in March 2020, the CDC was unable to provide testing kits for the first several weeks due to the contamination of tests at a manufacturing lab. Therefore, the government decided to permit diagnostic manufacturers to develop COVID-19 test kits and sell them to labs. They were also permitted to sell them to independent and hospital labs to develop LDTs under emergency use authorizations (EUAs). The primary benefit of an LDT over a typical diagnostic test is the time involved in the approval process. A routine diagnostic test can take years to pass through the many phases of research, testing, clinical evaluation, development of manufacturing processes, and review by the FDA or other regulatory authorities before a commercial test is available for public use. Whereas LDTs are designed to take only a few months to pass clearance and become regulated for use.
By late July 2020, LabCorp was processing nearly 180,000 tests for COVID-19 per day with an average turnaround time of three to five days. At the same time, Quest Diagnostics was performing 135,000 tests daily with an average wait of at least seven days.
President, CEO, and Chairman of LabCorp Adam H. Schechter, noted during their Q2 2020 earnings call how vital LDTs were in helping them keep up with the high demand for tests, “We launched back in March with our LDT, and it’s been remarkable the amount of volume we’ve been able to do through that LDT. We were doing 2,000 to 3,000 PCR tests per week, and here we are at the end of July, and we’re able to do 180,000 PCR tests per day. Of course, we still need our suppliers and the other companies to help us get to 180,000, but I have to say that our scientists did an extraordinary job with the LDT to get us to 180,000 today.”
When asked how many of the 180,000 tests were related to LDTs, Schechter said, “I’m not going to give you the exact number through the LDT because that changes, but we have 16 labs running tests. So, at the end of the day, it depends on where the samples are, which tests we run them, and what we try to do is optimize our network so we can get the best possible turnaround we can.”
Mark J. Guinan, VP, and CFO of Quest Diagnostics, also spoke on the positive impact of LDTs during their Q2 2020 earnings call.
“We exited the second quarter averaging approximately 110,000 and 26,000 COVID-19 molecular and serology tests, respectively, each day,” he said. “Over the next couple of weeks, we expect to have the capacity to perform 150,000 molecular diagnostic tests a day. We are providing test results in about two days for the highest priority patients. And the average turnaround time for nonpriority patients is at least seven days.”
Comparatively, before the pandemic the typical turnaround time for a lab test was one to two days.
LDTs Regulatory Landscape Continued
On August 19, 2020, the Department of Health and Human Services (HHS) Secretary Alex Azar announced HHS was rescinding all informal policy documents issued by the FDA related to LDTs because the FDA had not engaged in “notice-and-comment” rulemaking. The FDA had not required labs developing LDTs to submit tests for premarket review and had deprioritized the review of EUA requests for COVID-19 LDTs in favor of traditional, kit-based in vitro diagnostics (IVDs) from commercial manufacturers. As a result, such products were no longer required to secure a EUA or other non-emergency marketing authorization from the agency before being launched commercially. However, labs that chose to use LDTs without FDA premarket review or authorization would no longer be eligible for Public Readiness and Emergency Preparedness (PREP) Act coverage absent approval, clearance, or authorization and they would remain subject to regulation under CLIA.
According to HHS.gov, the PREP Act authorizes the secretary of the HHS to issue a declaration providing immunity from liability for claims; 1) of loss caused, arising out of, relating to, or resulting from administration or use of countermeasures to diseases, threats, and conditions, 2) determined by the secretary to constitute a present, or credible risk of a future public health emergency, 3) to entities and individuals involved in the development, manufacturing, testing, distribution, administration, and use of such countermeasures. Labs that already had an active EUA for LDTs were unaffected by the announcement.
With the pandemic appearing to slow down and commercial tests for COVID-19 becoming abundant, HHS hit the reset button for the regulation of LDTs on November 15, 2021. The FDA once again had regulatory authority and required clinical labs to submit EUA requests to continue performing and marketing such tests. The agency reported in their November 15 press release that the current areas of focus for EUA reviews were: 1) at-home and point-of-care (POC) diagnostic tests, 2) certain high-volume, lab-based molecular diagnostic tests, 3) certain lab-based and POC high-volume serology tests, and 4) tests submitted or supported by a U.S. government stakeholder.
The dilemma of the FDA’s authority to regulate LDTs has long been a complex and controversial topic for many decades. As mentioned previously, the FDA has historically delegated the oversight of LDTs to CLIA because they initially were relatively simple lab tests and available on a limited basis. However, due to technological advances, LDTs have evolved and are now much more complex with a nationwide reach and present higher risks. The FDA’s main argument is inaccurate test results could lead to people initiating unnecessary treatment, delaying treatment, or foregoing treatment for a health condition that could result in severe illness or death. Whereas most clinical labs feel that LDTs are already sufficiently regulated under CLIA and should not be double regulated under the FDA.
Professor of Pathology and Immunology at Washington University Dennis Dietzen, Ph.D., said, “LDTs fill a void where there is no FDA-approved test. We need to be able to build these things with a healthy amount of regulation, but not a burdensome amount. If labs had to go through premarket review for all their LDTs, it would make the assays more expensive to run, and I think a lot of laboratories would just throw in the towel.” (9) (13) (14) (15) (16) (17) (18) (19)
Conclusion
When COVID-19 initially hit the U.S. in March 2020, the CDC was unable to provide testing kits for the first several weeks due to the contamination of tests at a manufacturing lab. Therefore, the government decided to permit diagnostic manufacturers to develop COVID-19 test kits and sell them to labs. They were also permitted to sell them to independent and hospital labs to develop LDTs under emergency use authorizations (EUAs). The primary benefit of an LDT over a typical diagnostic test is the time involved in the approval process. A routine diagnostic test can take years to pass through the many phases of research, testing, clinical evaluation, development of manufacturing processes, and review by the FDA or other regulatory authorities before a commercial test is available for public use. Whereas LDTs are designed to take only a few months to pass clearance and become regulated for use.
By late July 2020, LabCorp was processing nearly 180,000 tests for COVID-19 per day with an average turnaround time of three to five days. At the same time, Quest Diagnostics was performing 135,000 tests daily with an average wait of at least seven days.
President, CEO, and Chairman of LabCorp Adam H. Schechter, noted during their Q2 2020 earnings call how vital LDTs were in helping them keep up with the high demand for tests, “We launched back in March with our LDT, and it’s been remarkable the amount of volume we’ve been able to do through that LDT. We were doing 2,000 to 3,000 PCR tests per week, and here we are at the end of July, and we’re able to do 180,000 PCR tests per day. Of course, we still need our suppliers and the other companies to help us get to 180,000, but I have to say that our scientists did an extraordinary job with the LDT to get us to 180,000 today.”
Sources:
- https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA
- Laboratory-Developed Tests (LDTs) – Testing.com
- Putting New Laboratory Tests into Practice – Testing.com
- https://www.fda.gov/medical-devices/ivd-regulatory-assistance/clinical-laboratory-improvement-amendments-clia
- QSO-20-21-CLIA (cms.gov)
- Impact of COVID-19 Pandemic on Laboratory Utilization – PubMed (nih.gov)
- Strengthening Clinical Laboratories | CDC
- CDC COVID Data Tracker: Home
- About Face: Laboratory-Developed Tests for COVID-19 Now Subject to EUA Requirements | Mintz
- ‘The challenges that labs are facing are complex’: Why COVID-19 test results are so delayed (nbcnews.com)
- Laboratory Corporation of America Holdings, Q2 2020 Earnings Call, Jul 28, 2020.pdf
- Quest Diagnostics Incorporated, Q2 2020 Earnings Call, Jul 23, 2020.pdf
- COVID-19 and Lab Testing: What’s the Story Behind the Story? | Mintz
- HHS Reverses Course on LDTs: COVID-19 LDTs Again Require FDA Premarket Review | Ropes & Gray LLP (ropesgray.com)
- Laboratory Developed Tests | FDA
- HHS Prohibits FDA from Requiring Premarket Review of LDTs, Including During the COVID-19 Emergency | Ropes & Gray LLP (ropesgray.com)
- Rescission of Guidances and Other Informal Issuances | HHS.gov (archive.org)
- Public Readiness and Emergency Preparedness Act (hhs.gov)
- Back to the Future on Laboratory Developed Tests | AACC.org
- Future of LDT Regulation Hangs in the Balance | AACC.org





